Vegetos

(An International Journal of Plant Research & Biotechnology)
(eISSN: 2229-4473)
In silico analysis of plumbagin against cyclin-dependent kinases receptor
Research Articles | Published: 11 January, 2021
First Page: 50
Last Page: 56
Views: 4367
Keywords: Cancer, Plumbagin, CDK6, Molecular docking, Dynamics
Abstract
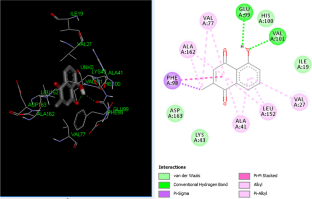
Cancer is an abnormal growth of cells which could migrate from its place of origin to other parts of the body. It is one of the most serious disease on which widespread research work has been going on. Computer aided drug designing has helped in the identification of potential leads that can be used for the development of a drug. Plumbagin is a naphthoquinone derivative from Plumbago zeylanica roots which possess strong anticancer properties. Plumbagin has been shown to produce inhibitory effects on multiple cancer-signaling proteins. However, binding mechanism and molecular interactions have not been elucidated yet for most of the target proteins. In this investigation, an attempt was done to explore the binding mechanism of plumbagin against cyclin-dependent kinases (CDK) receptor using molecular docking. The least binding energy of plumbagin with CDK6 was found to be − 6.18 kcal/mol. The molecular simulation suggests that plumbagin has potential binding affinities with CDK6 and its interactions with CDK6 was quite stable during the whole period of simulation run. It was also found that plumbagin obeys Lipinski’s Rule of 5 and has drug likeness proved by ADMET analysis. As plumbagin is a natural compound, it has reduced side effects and these results would be useful for cancer treatment.
References
- Agirre X, Vilas-Zornoza A, Jiménez-Velasco A, Martin-Subero JI, Cordeu L, Gárate L et al (2009) Epigenetic *silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Can Res 69(10):4443–4453
- Aleem E, Arceci RJ (2015) Targeting cell cycle regulators in hematologic malignancies. Front Cell Dev Biol 3:16
- Bertoli C, Skotheim JM, De Bruin RA (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14(8):518–528
- Cho YS, Borland M, Brain C, Chen CH, Cheng H, Chopra R, Chung K, Groarke J, He G, Hou Y, Kim S, Kovats S, Lu Y, O’Reilly M, Shen J, Smith T, Trakshel G, Vögtle M, Xu M, Xu M et al (2010) 4-(Pyrazol-4-yl)-pyrimidines as selective inhibitors of cyclin-dependent kinase 4/6. J Med Chem 53(22):7938–7957
- Davies TG, Pratt DJ, Endicott JA, Johnson LN, Noble ME (2002) Structure-based design of cyclin-dependent kinase inhibitors. Pharmacol Ther 93(2–3):125–133
- Grover A, Singh R, Shandilya A, Priyandoko D, Agrawal V, Bisaria VS et al (2012) Ashwagandha derived withanone targets TPX2-Aurora A complex: computational and experimental evidence to its anticancer activity. PLoS ONE 7(1):e30890
- Hernandez Maganhi S, Jensen P, Caracelli I, Zukerman Schpector J, Fröhling S, Friedman R (2017) Palbociclib can overcome mutations in cyclin dependent kinase 6 that break hydrogen bonds between the drug and the protein. Protein Sci 26(4):870–879
- Jamal MS, Parveen S, Beg MA, Suhail M, Chaudhary AG, Damanhouri GA et al (2014) Anticancer compound plumbagin and its molecular targets: a structural insight into the inhibitory mechanisms using computational approaches. PLoS ONE 9(2):e87309
- Jayaraman A, Jamil K (2014) Drug targets for cell cycle dysregulators in leukemogenesis: in silico docking studies. PLoS ONE 9(1):e86310
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90
- Kawasaki Y, Komiya M, Matsumura K, Negishi L, Suda S, Okuno M et al (2016) MYU, a target lncRNA for Wnt/c-Myc signaling, mediates induction of CDK6 to promote cell cycle progression. Cell Rep 16(10):2554–2564
- Li Y, Zhang J, Gao W, Zhang L, Pan Y, Zhang S, Wang Y (2015) Insights on structural characteristics and ligand binding mechanisms of CDK2. Int J Mol Sci 16(5):9314–9340
- Liu X, Feng J, Tang L, Liao L, Xu Q, Zhu S (2015) The regulation and function of miR-21-FOXO3a-miR-34b/c signaling in breast cancer. Int J Mol Sci 16(2):3148–3162
- Molassiotis A, Panteli V, Patiraki E, Ozden G, Platin N, Madsen E et al (2006) Complementary and alternative medicine use in lung cancer patients in eight European countries. Complement Ther Clin Pract 12(1):34–39
- Roy A, Ahuja S, Bharadvaja N (2017) A review on medicinal plants against cancer. J Plant Sci Agric Res 2:008
- Roy A, Bharadvaja N (2017b) Establishment of the shoot and callus culture of an important medicinal plant Plumbago zeylanica. Adv Plants Agric Res 7(5):00274
- Roy A, Bharadvaja N (2017a) Medicinal plants in the management of cancer: a review. Int J Complement Alt Med 9(2):00291
- Roy A, Bharadvaja N (2018a) Effect of various culture conditions on shoot multiplication and GC–MS analysis of Plumbago zeylanica accessions for plumbagin production. Acta Physiol Plant 40(11):190
- Roy A, Bharadvaja N (2018b) Biotechnological approaches for the production of pharmaceutically important compound: plumbagin. Curr Pharm Biotechnol 19(5):372–381
- Roy A, Jauhari N, Bharadvaja N (2018) Medicinal plants as a potential source of chemopreventive agents. In: Akhtar MS, Swamy MK (eds) Anticancer plants: natural products and biotechnological implements. Springer, Singapore, pp 109–139
- Sakpakdeejaroen I, Somani S, Laskar P, Mullin M, Dufès C (2019) Transferrin-bearing liposomes entrapping plumbagin for targeted cancer therapy. Journal of Interdisciplinary Nanomedicine 4(2):54–71
- Schulze-Gahmen U, Kim SH (2002) Structural basis for CDK6 activation by a virus-encoded cyclin. Nature Structural Biology 9(3):177–181
- Sliwoski G, Kothiwale S, Meiler J, Lowe EW (2014) Computational methods in drug discovery. Pharmacol Rev 66(1):334–395
- Tadesse S, Yu M, Kumarasiri M, Le BT, Wang S (2015) Targeting CDK6 in cancer: state of the art and new insights. Cell Cycle 14(20):3220–3230
- Tripathi SK, Panda M, Biswal BK (2019) Emerging role of plumbagin: cytotoxic potential and pharmaceutical relevance towards cancer therapy. Food Chem Toxicol 125:566–582
- Wipf P, Skoda EM, Mann A (2015) Conformational restriction and steric hindrance in medicinal chemistry. In: Wermuth CG, Aldous D, Raboisson P, Rognan D (eds) The practice of medicinal chemistry. Academic Press, Burlington, pp 279–299
- Zhang J, Zhang L, Xu Y, Jiang S, Shao Y (2018) Deciphering the binding behavior of flavonoids to the cyclin dependent kinase 6/cyclin D complex. PLoS ONE 13(5):e0196651
Author Information
Department of Biotechnology, School of Engineering & Technology, Sharda University, Greater Noida, India